混池测序分析(Bulked segregant analysis)是一种省时省力、广泛应用于挖掘性状遗传位点的方法,已有多种相应算法被开发出来。包括基于高低池等位基因频率差异的ΔSNP-index方法,基于欧式距离的ED4方法,基于G值计算的G’方法,基于LOD值计算的SmoothLOD方法以及基于非参数检验的Ridit方法等。这些方法缺乏用户友好型软件,从而使用复杂;同时大多只适用于两个混池,且难以检测复杂性状和背景下的微效位点。
近日,华中农业大学植物科学技术学院李林教授课题组和理学院陈洪教授课题组联合在Molecular Plant发表了题为“DeepBSA: A deep-learning algorithm improves bulked segregant analysis for dissecting complex traits”的研究论文,报道了利用深度学习进行功能基因定位的混池测序新算法。
该研究首先设计了一套分级混池的混池测序方案,再利用玉米株高的混池测序数据,构建了一个残差连接的U-Net深度学习模型。训练学习后,模型在2-10个不同数目混池的AUC值在0.87-0.96之间。进一步通过生成仿真数据,来测试不同效应位点、不同群体大小、不同测序深度及不同混池数等条件下的定位效率,发现DeepBSA能高效鉴定到各复杂性状下的所有功能位点。同时,将此算法与五种广泛应用的算法进行比较,证明此算法定位的结果准确度更高,噪音更小。
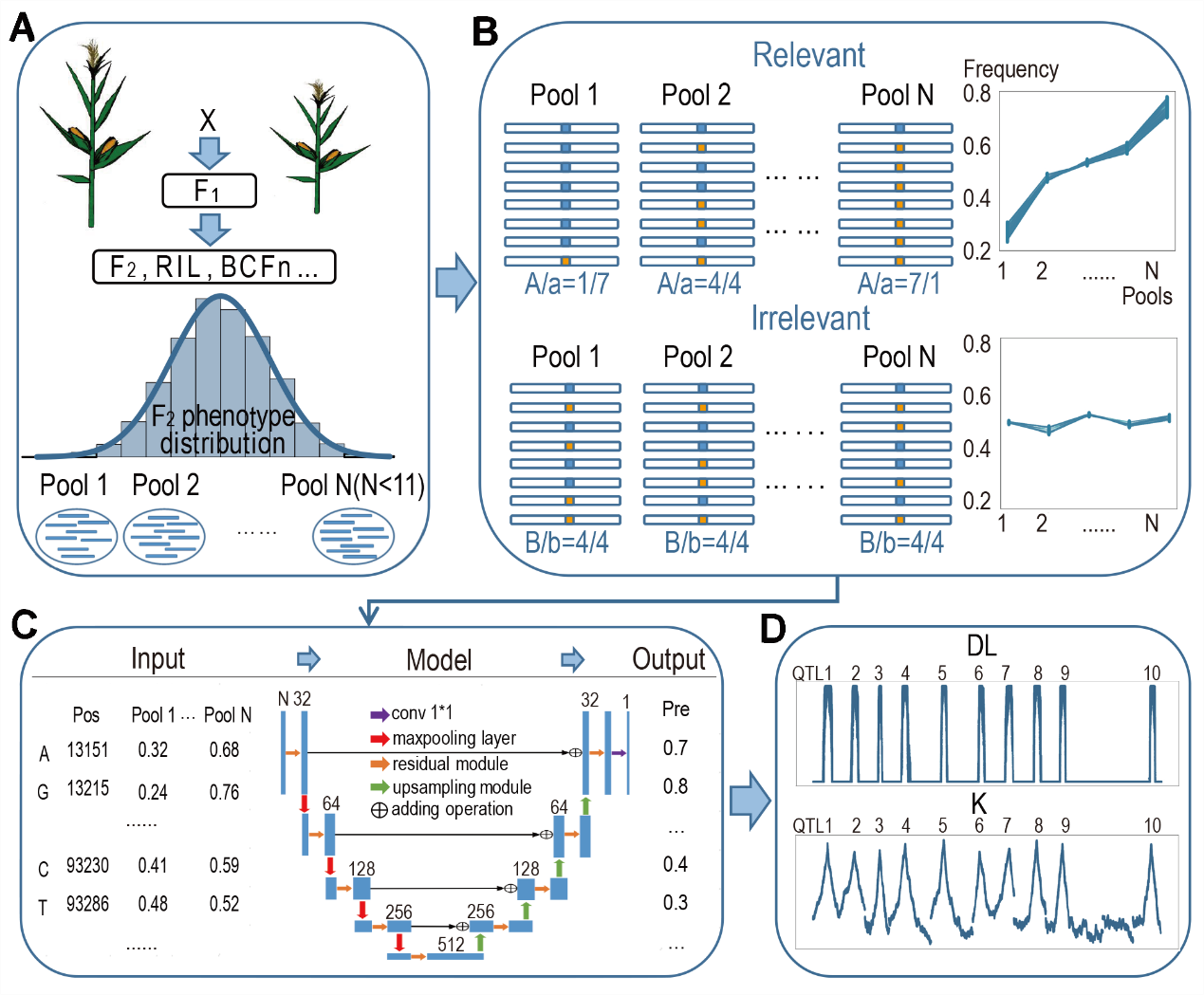
图1. DeepBSA原理流程
通过对不同混池数的公共数据进行分析,DeepBSA不仅能鉴定到原方法鉴定的结果,还能找到新的潜在功能位点。同时,该方法对物种及性状有着广泛的适应性,其在水稻株高和开花期、玉米株高及武昌鱼肌间刺等性状中均得到良好的鉴定效果。因此,DeepBSA在动植物复杂性状的功能位点克隆方面具有重要应用前景。

图2. DeepBSA的操作界面及结果展示
之前发表的混池测序算法对使用者都有一定的生物信息学要求,操作相对复杂。因此该研究将开发的新算法和五种广泛使用的算法集成为一个软件,包括数据预处理、功能位点定位以及模拟数据生成等功能。同时开发出一个用户友好的操作界面(http://zeasystemsbio.hzau.edu.cn/tools.html),通过简单操作可以快速获得定位的图表,还可以比较不同方法的结果差异。
除了提供一种应用广泛且更精准的混池测序新算法,研究者相信开发的用户友好界面能帮助众多科研工作者简单快速得到混池测序定位结果,从而加速动植物复杂性状的功能位点克隆及解析。
华中农业大学作物遗传改良全国重点实验室和湖北洪山实验室的李林教授和农业生物信息湖北省重点实验室李伟夫副教授为论文通讯作者。华中农业大学博士研究生李昭和硕士研究生陈晓轩为论文第一作者。华中农业大学陈洪教授、已毕业硕士研究生施韶强、博士研究生王席和中国农科院作科所张红伟副研究员也参与了该研究。该研究得到国家自然科学基金、海南崖州湾种子实验室和湖北洪山实验室重大项目等经费的支持。
【英文摘要】
Bulked segregant analysis (BSA) is a rapid, cost-effective method for mapping mutations and quantitative trait loci (QTLs) in animals and plants based on high-throughput sequencing. However, the algorithms currently used for BSA have not been systematically evaluated and are complex and fallible to operate. We developed a BSA method driven by deep learning (DL) —DeepBSA for QTL mapping and functional gene cloning. DeepBSA is compatible with a variable number of bulked pools and performed well with various simulated and real datasets in both animals and plants. DeepBSA outperformed all other algorithms when comparing absolute bias and signal-noise-ratio. Moreover, we applied DeepBSA to an F2 segregating maize population of 7,160 individuals and uncovered five candidate QTLs, including three well-known plant-height genes. Finally, we developed a user-friendly graphical user interface (GUI) for DeepBSA, integrating five widely used BSA algorithms and our two newly developed algorithms, which is easy to operate and can quickly map QTLs and functional genes. The DeepBSA software is publicly available at http://zeasystemsbio.hzau.edu.cn/tools.html.
论文链接:https://www.cell.com/molecular-plant/fulltext/S1674-2052(22)00267-2
